Zulassung von Medizinprodukten
Die Zulassung von Medizinprodukten in Europa wird durch die Medical Device Regulation (MDR) geregelt. Diese Verordnung stellt sicher, dass alle Medizinprodukte, die auf den Markt kommen, höchsten Standards in Bezug auf Sicherheit, Qualität und Wirksamkeit entsprechen.
Im Rahmen der MDR müssen Hersteller umfangreiche Anforderungen erfüllen, darunter die Erstellung einer vollständigen technischen Dokumentation, eine klinische Bewertung zur Überprüfung der Wirksamkeit, Nachweise zur Biokompatibilität, ein umfassendes Risikomanagement und die Sicherstellung der Gebrauchstauglichkeit.
Diese Anforderungen sind entscheidend, um das Vertrauen der Anwender zu stärken und die Patientensicherheit zu gewährleisten. Die MDR legt außerdem großen Wert auf die kontinuierliche Überwachung von Medizinprodukten, um deren Sicherheit auch nach der Zulassung sicherzustellen.
Hopp + Flaig steht Ihnen als kompetenter Partner zur Seite und unterstützt Sie bei der Einhaltung aller MDR-Vorgaben – von der Vorbereitung der technischen Dokumentation bis hin zur erfolgreichen Marktzulassung.
-
MDR
-
Technische Dokumentation
-
Klinische Bewertung
-
Biokompatibilität
-
Gebrauchstauglichkeit
-
Risikomanagement
-
Prozessvalidierung
-
MDSAP
-
FDA
-
IVDR
-
Großhandelserlaubnis
MDR Beratung für Medizinproduktehersteller
Die Zulassung von Medizinprodukten gemäß MDR ist ein wichtiger Prozess, um sicherzustellen, dass Produkte den strengen EU-Standards für Sicherheit, Qualität und Rückverfolgbarkeit entsprechen. Die Medical Device Regulation (MDR), oft auch als MDR Verordnung bezeichnet, fordert, dass alle Medizinprodukte, die in der EU vertrieben werden, eine gründliche Konformitätsbewertung durchlaufen und die CE-Kennzeichnung erhalten müssen. Die CE-Kennzeichnung zeigt, dass Produkte die Anforderungen der CE-Richtlinie erfüllen und für den europäischen Markt zugelassen sind. Hierzu gehören auch eine präzise technische Dokumentation für Medizinprodukte, eine fundierte klinische Bewertung und ein umfassendes Risikomanagement.
Unser Team aus MDR-Beratern, Auditoren und Regulatory-Experten unterstützt Sie in jeder Phase des MDR-Zulassungsprozesses – von der Gap-Analyse über die technische Dokumentation bis zur finalen Audit-Prüfung durch die benannte Stelle.
Wie wir Sie dabei unterstützen:
- Strukturierung: Wir analysieren Ihre Produktakten, um festzustellen, welche Dokumente und Nachweise für die Zulassung von Medizinprodukten gemäß MDR erforderlich sind. Durch eine präzise Strukturierung Ihrer technischen Dokumentation schaffen wir eine solide Grundlage für den weiteren Zertifizierungsprozess.
- Individuelle Maßnahmenplanung: Ein detaillierter Maßnahmenplan ist der Schlüssel zur erfolgreichen MDR-Zulassung. Unser Plan deckt alle Schritte ab, die zur MDR-Konformität führen und berücksichtigt die spezifischen Anforderungen Ihrer medizinischen Produkte.
- Begleitung bei der Umsetzung: Während der Umsetzungsphase unterstützen wir Sie aktiv bei der Einhaltung aller MDR-Vorgaben und der CE-Kennzeichnungsanforderungen.
- Dokumentation: Eine vollständige technische Dokumentation ist entscheidend für die MDR-Compliance und die CE-Zertifizierung. Wir helfen Ihnen bei der Erstellung und Pflege Ihrer Unterlagen, sodass Sie bestens auf Audits und Prüfungen der Benannten Stellen vorbereitet sind.
Warum mit uns?
- Erfahrene MDR-Berater, Auditoren & Zertifizierungsexperten
- Individuelle MDR-Strategien für Medizinprodukte-Hersteller
- Sichere & effiziente Zulassung gemäß Medical Device Regulation
Starten Sie jetzt Ihre MDR-Zulassung mit unseren Experten für Audit & Zertifizierung. Kontaktieren Sie uns für eine unverbindliche Erstberatung.
Der Nachweis für Ihr konformes Medizinprodukt
Mit der Einführung der MDR sind die Anforderungen an die technische Dokumentation deutlich gestiegen. Die technische Dokumentation ist gemäß MDR-Anhang II das zentrale Nachweisinstrument für Ihr Medizinprodukt. Sie enthält alle wichtigen Informationen zu Konstruktion, Herstellung und Funktionsweise und zeigt, dass Ihr Produkt alle regulatorischen Anforderungen und länderspezifischen Vorschriften erfüllt. Diese Dokumentation ist die Grundlage für die Konformitätserklärung und CE-Kennzeichnung und muss auf Anforderung jederzeit den Überwachungsbehörden vorgelegt werden können. Viele Hersteller stehen vor Herausforderungen wie beispielsweise die Bereitstellung von detaillierten klinischen Nachweisen und der kontinuierlichen Aktualisierung der Dokumentation.
Unsere Beratung zur technischen Dokumentation MDR
Wir unterstützen Sie umfassend mit unserer Beratung zur technischen Dokumentation MDR, um sicherzustellen, dass Ihre Dokumentation den gesetzlichen Anforderungen entspricht. Durch unsere langjährige Erfahrung als Dienstleister für technische Dokumentation MDR helfen wir Ihnen, eine vollständige und konforme Dokumentation zu erstellen.
Wir unterstützen Sie dabei:
- Analyse Technische Dokumentation MDR: Wir prüfen Ihre bestehenden Dokumente umfassend und identifizieren gezielt Lücken sowie Optimierungsbedarf, um sicherzustellen, dass Ihre technische Dokumentation allen Anforderungen der MDR entspricht.
- Erstellung und Prozesse Technische Dokumentation MDR: Ob Sie eine vollständige technische Dokumentation erstellen oder gezielte Erweiterungen vornehmen möchten – unser Team übernimmt die Dokumentation für Sie und unterstützt Sie bei der Implementierung der erforderlichen Prozesse Technische Dokumentation MDR.
- Kontinuierliche Überwachung nach Inverkehrbringen: Wir unterstützen Sie bei der Implementierung eines Post-Market Surveillance (PMS)-Systems gemäß MDR Anhang III. Dies umfasst das Post-Market Clinical Follow-up (PMCF) zur kontinuierlichen Aktualisierung der klinischen Bewertung sowie die Vigilanz zur Erfassung und Meldung von Vorkommnissen und Trends zur frühzeitigen.
Warum unsere Expertise entscheidend für Ihre Technische Dokumentation MDR ist
Verlassen Sie sich auf unsere umfassende Projekterfahrung und Expertise, um Ihre technische Dokumentation MDR-konform zu halten. Wir verstehen die komplexen Anforderungen und Hürden, die Hersteller oft durch die MDR meistern müssen, und helfen Ihnen, alle Dokumentationspflichten effizient zu erfüllen. So können Sie sich auf Ihre Kernkompetenzen konzentrieren.
Beratung Klinische Bewertung nach MDR
Unsere Beratung klinische Bewertung MDR stellt sicher, dass Ihr Medizinprodukt die regulatorischen Anforderungen der Verordnung (EU) 2017/745 (MDR) erfüllt. Die klinische Bewertung ist ein essenzieller Bestandteil des Konformitätsbewertungsverfahrens und der technischen Dokumentation. Der Nachweis für die Sicherheit und Leistungsfähigkeit Ihres Medizinproduktes erfolgt anhand von vorhandenen klinischen Daten. Für innovative oder spezielle Produkte ohne ausreichende Vergleichsdaten ist hingegen eine klinische Studie notwendig, um die erforderlichen Nachweise zu liefern.
Wir begleiten Sie Schritt für Schritt und unterstützen Sie gezielt bei der Einführung der klinischen Bewertung nach MDR, der Analyse bestehender klinischer Daten sowie der Erstellung einer umfassenden Dokumentation für bestehende und vergleichbare Produkte auf dem Markt.
Wir unterstützen Sie dabei:
- Klinischer Entwicklungsplan: Gemeinsam entwickeln wir eine fundierte Strategie zur Sammlung und Dokumentation aller notwendigen klinischen Daten – die Grundlage für eine zielgerichtete und effiziente klinische Bewertung.
- Analyse klinische Bewertung MDR: Wir prüfen vorhandene klinische Daten und führen eine GAP-Analyse der klinischen Bewertung nach MDR durch. Dies hilft, Schwachstellen zu identifizieren und ein fundiertes Nutzen-Risiko-Profil zu erstellen.
- Klinischer Evaluierungsbericht: Wir erstellen einen umfassenden Evaluierungsbericht, der alle Ergebnisse und Bewertungen dokumentiert.
- Kontinuierliche Überwachung: Wir unterstützen Sie bei der Planung und Durchführung von Post-Market Clinical Follow-Up (PMCF)-Maßnahmen, um die langfristige Sicherheit und Leistungsfähigkeit Ihres Produkts sicherzustellen.
Nutzen Sie unsere Beratung zur klinischen Bewertung nach MDR und sichern Sie den erfolgreichen Marktzugang Ihres Medizinprodukts!
Beratung & Beurteilung der Biokompatibilität gemäß DIN EN ISO 10993
Die Beratung zur Beurteilung der Biokompatibilität von Medizinprodukten ist entscheidend, um gesundheitliche Risiken zu reduzieren und die Verträglichkeit im menschlichen Körper zu gewährleisten. Die Biokompatibilitätsprüfung erfolgt gemäß DIN EN ISO 10993 durch biologische Tests und die Analyse potenzieller chemischer Stoffe, die das Material freisetzen könnte.
Unsere GAP-Analyse zur Biokompatibilität (MDR) hilft Ihnen, bestehende Prüfprozesse zu bewerten und notwendige Maßnahmen zur Einhaltung der regulatorischen Anforderungen abzuleiten. Dabei greifen wir auf eine Vielzahl an Daten zurück – von technischen Spezifikationen über Materialanalysen bis hin zu Erfahrungswerten vergleichbarer Produkte. Wir berücksichtigen dabei sowohl das Material selbst als auch Faktoren wie Materialveränderungen über den gesamten Lebenszyklus, Produktionsbearbeitungen, Endreinigung und den Einsatz von Lösungsmitteln. Auch die Wechselwirkungen zwischen verschiedenen Materialien und die Auswirkungen einer wiederholten Anwendung sind zentrale Aspekte unserer Biokompatibilitätsanalyse.
Wir unterstützen Sie dabei:
- Beratung zur Auswahl von Biokompatibilitätstests: Wir stehen Ihnen zur Seite, wenn es um die Auswahl geeigneter Tests geht – von Zytotoxizität (ISO 10993-5) über Sensibilisierung (ISO 10993-10) und Irritation (ISO 10993-23) bis hin zu Hämokompatibilitätstests (ISO 10993-4).
- Bewertung der Testergebnisse: Nutzen Sie unsere Expertise für eine fundierte Analyse Ihrer Biokompatibilitätsergebnisse. Wir unterstützen Sie dabei, alle nötigen Schritte zur sicheren Umsetzung und Dokumentation der biologischen Sicherheit zu meistern.
- Zusammenarbeit mit akkreditierten Laboren: Vertrauen Sie auf unsere Verbindungen zu zertifizierten Prüflaboren. Wir übernehmen die gesamte Koordination, sodass Ihre Prüfungen reibungslos ablaufen.
Unser erfahrenes Team begleitet Sie durch den gesamten Prozess der biologischen Beurteilung, um sicherzustellen, dass Ihre Medizinprodukte alle Sicherheits- und Qualitätsstandards der DIN EN ISO 10993 erfüllen.
Beratung Gebrauchstauglichkeit MDR
Die Gebrauchstauglichkeit (Usability) für Medizinprodukte ist dazu da, um Benutzungsfehler zu vermeiden und damit zur Sicherheit des Medizinprodukts beizutragen. Ist die Gebrauchstauglichkeit unzureichend, kann dies zu erheblichen Schäden oder sogar zum Tod des Patienten führen.
Die EN 62366-1 spielt eng mit der ISO 13485 zusammen und beschreibt einen „Usability Engineering Prozess“, der ein vertretbares Anwendungsrisiko für Medizinprodukte durch einfache Bedienung und Benutzung sicherstellen soll. Dabei soll die ganze Lebensdauer inklusive Transport, Lagerung, Installation, Betrieb, Wartung, Reparatur sowie die Entsorgung miteingebzogen werden. Im Prozess werden Maßnahmen zur Risikobeherrschung in Bezug auf Design, Nutzerschnittstelle und die Bereitstellung von Sicherheitsinformationen beschrieben.
Mit unsere Beratung Gebrauchstauglichkeit MDR begleiten Sie durch den gesamten Prozess für eine normgerechte Gebrauchstauglichkeit nach IEC 62366-1.
Wir unterstützen Sie dabei:
- Spezifikation der Gebrauchstauglichkeit: Wir entwickeln präzise Anforderungsspezifikationen, die den realen Einsatzbereich und die Bedürfnisse Ihrer Zielgruppe berücksichtigen.
- Berichterstellung nach Usability-Tests: Wir erstellen aussagekräftige Berichte zu Gebrauchstauglichkeitsprüfungen, die die Sicherheit und Benutzerfreundlichkeit Ihrer Produkte nachweisen.
- Erstellung der Gebrauchstauglichkeitsakte: Unsere Experten erstellen vollständige, normgerechte Dokumentationen gemäß IEC 62366-1 und IEC TR 62366-2.
- Prüfung und Optimierung bestehender Gebrauchstauglichkeitsakten: Wir analysieren und optimieren Ihre bestehenden Dokumente, um Vollständigkeit, Normkonformität und Qualität zu gewährleisten.
Mit unserer Beratung Gebrauchstauglichkeit MDR helfen wir Ihnen, die Anforderungen an sichere und normgerechte Medizinprodukte zu erfüllen. Vertrauen Sie auf unsere Erfahrung, um die Anforderungen der IEC 62366-1 und IEC TR 62366-2 erfolgreich umzusetzen.
Beratung ISO 14971 Risikomanagement
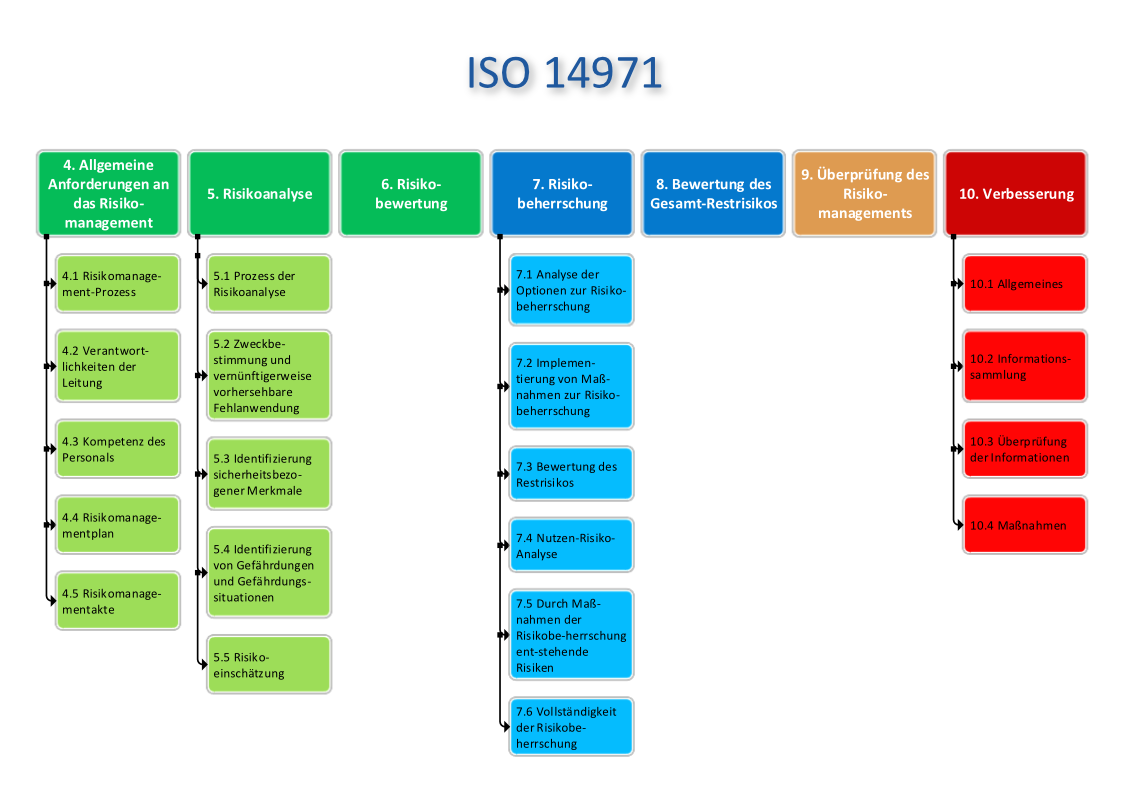
Das Risikomanagement für Medizinprodukte, geregelt durch die DIN EN ISO 14971, bildet die Grundlage für die Identifikation, Bewertung und Kontrolle von Risiken entlang des gesamten Produktlebenszyklus. Ob Entwicklung, Produktion oder Marktüberwachung – ein wirksames Risikomanagementsystem optimiert Prozesse und Produkte und trägt somit zur Sicherheit von Patienten bei. Nebenbei gewährleistet man die Einhaltung regulatorischer Anforderungen wie des Qualitätsmanagements ISO 13485 und der MDR.
Warum ist das Risikomanagement entscheidend?
Ein strukturiertes ISO 14971 Risikomanagement minimiert Gefahren, verbessert die Produktsicherheit und trägt zur Risikominimierung bei. Mit der richtigen Anwendung von Risikomanagement-Methoden lassen sich Risiken identifizieren, bewerten und minimieren sowie eine effektive Risikobewertung-Analyse durchführen. Diese Norm fordert zudem eine sorgfältige Dokumentation – von der Planung über die Durchführung bis zur Pflege der Risikomanagementakte gemäß ISO 14971 – um Prüfungen durch benannte Stellen erfolgreich zu bestehen.
Darüber hinaus ist das Risikomanagement ein zentraler Bestandteil der Technischen Dokumentation gemäß MDR Anhang II. Die Anforderungen der MDR unterstreichen die Bedeutung einer umfassenden und genauen Dokumentation, die sowohl die Sicherheit als auch die Einhaltung regulatorischer Vorgaben gewährleistet. Eine strukturierte Einführung der ISO 14971 stellt sicher, dass alle relevanten Prozesse normkonform umgesetzt werden.
Wir unterstützen Sie dabei:
- Einführung ISO 14971: Wir begleiten Sie durch den gesamten Implementierungsprozess und sorgen für eine regelkonforme Umsetzung.
- Erstellung von Risikomanagementplänen gemäß ISO 14971: Wir definieren Maßnahmen zur effektiven Risikobewältigung.
- Durchführung von Risikoanalysen: Mit bewährten Methoden wie der FMEA identifizieren wir potenzielle Gefährdungen, definieren Maßnahmen zur Risikominimierung oder -vermeidung und bewerten verbleibende Restrisiken.
- Pflege & Analyse Ihrer Risikomanagementakte: Wir sorgen für die regelmäßige Aktualisierung und lückenlose Dokumentation Ihrer Risikomanagement-Berichte.
- Bereitstellung von Vorlagen: Wir stellen praxisbewährte Vorlagen für Risikoanalysen und Risikobewertungsmatrizen zur Verfügung.
Mit unserer umfassenden Erfahrung in der Risikobewertung von Medizinprodukten bieten wir praxisorientierte und normkonforme Lösungen für Ihre individuelle Situation. Wir stellen sicher, dass Ihr Prozess des Risikomanagements effektiv und effizient umgesetzt wird. Wir unterstützen Sie bei jedem Schritt Ihres Prozesses des Risikomanagements. Kontaktieren Sie uns für eine unverbindliche Beratung.
Normenanforderung
Beratung zur Prozessvalidierung MDR
Prozessvalidierung bedeutet, sicherzustellen und Nachweise zu erbringen, dass (Produktions-) Prozesse den festgelegten Anforderungen entsprechen und zuverlässig für den vorgesehenen Zweck geeignet sind. Eine Validierung muss immer dann durchgeführt werden, wenn sich das Prozessergebnis nicht verifizieren lässt. Die Prozessvalidierung ist ein zentraler Bestandteil des Qualitätsmanagements gemäß ISO 13485 und stellt sicher, dass Produkte höchsten Sicherheits- und Qualitätsstandards gerecht werden.
ISO 13485 fordert, dass Produktionsprozesse korrekt entwickelt, implementiert und über den gesamten Lebenszyklus hinweg konsistente, wiederholbare und reproduzierbare Ergebnisse liefern. Dies ist ein wesentlicher Schritt, um die Produktsicherheit, Wirksamkeit und Konformität Ihrer Medizinprodukte sicherzustellen. Kritische Prozesse, die nicht durch nachträgliche Prüfungen überprüft werden können, erfordern eine detaillierte GAP-Analyse der Prozessvalidierung, um potenzielle Lücken in der Validierungsstrategie zu identifizieren und zu schließen.
Die Prozessvalidierung erfolgt in fünf Qualifizierungsphasen:
- PVD (Process Validation Decision): Im ersten Schritt wird entschieden, welche Prozesse validiert werden müssen. Dabei wird definiert, welche Anforderungen der Prozess erfüllen muss, um das Medizinprodukt sicher und zuverlässig herzustellen.
- DQ (Design Qualification): Hier wird geprüft, welche Anlagen für den Herstellungsprozess benötigt werden. Außerdem wird sichergestellt, dass diese alle Anforderungen erfüllen, um den Prozess wie geplant durchzuführen.
- IQ (Installationsqualifizierung): In dieser Phase wird dokumentiert, dass alle Anlagen korrekt installiert wurden und den technischen Spezifikationen entsprechen. Auch wird geprüft, ob die Vorgaben der Gerätehersteller eingehalten wurden.
- OQ (Funktionsqualifizierung): Es wird überprüft, ob die Prozessparameter, wie z. B. Kontroll- und Eingreifgrenzen, unter definierten Bedingungen zuverlässig ein Produkt erzeugen, das alle Qualitätsanforderungen erfüllt.
- PQ (Leistungsqualifizierung): Hier wird nachgewiesen, dass der Prozess unter realen Produktionsbedingungen – einschließlich Worst-Case-Szenarien – dauerhaft Produkte liefert, die den festgelegten Anforderungen entsprechen.
Wie wir Sie dabei unterstützen:
- Beratung zur Prozessvalidierung (MDR): Wir begleiten Sie bei der Einführung der Prozessvalidierung (MDR) und der Umsetzung der regulatorischen Anforderungen.
- Durchführung einer GAP-Analyse Prozessvalidierung: Identifikation von Schwachstellen und Optimierung der Validierungsstrategie.
- Erstellung eines Validierungsmasterplans: Wir entwickeln einen umfassenden Plan, der Validierungsziele definiert, alle Schritte klar strukturiert und die Revalidierungsfrequenz festlegt, um eine reibungslose und nachhaltige Umsetzung sicherzustellen.
- Moderation von Besprechungen: Wir moderieren interdisziplinäre Besprechungen, um die Zusammenarbeit zwischen Abteilungen zu fördern und alle Projektbeteiligten optimal einzubinden.
- Dokumentation Prozessvalidierung: Wir erstellen normgerechte Validierungsberichte, die den gesamten Validierungsprozess transparent und nachvollziehbar dokumentieren.
- Schulung Ihrer Mitarbeiter: Wir schulen Ihre Teams gezielt, damit alle Beteiligten bestens auf ihre Aufgaben vorbereitet sind und den Anforderungen gerecht werden.
Expertenunterstützung für eine erfolgreiche Prozessvalidierung
Ob es um die Einführung der Prozessvalidierung (MDR), eine GAP-Analyse der Prozessvalidierung, die Zertifizierung der Prozessvalidierung oder die vollständige Dokumentation der Prozessvalidierung geht – wir stehen Ihnen als kompetenter Partner zur Seite. Kontaktieren Sie uns und profitieren Sie von unserer langjährigen Erfahrung in der Beratung zur Prozessvalidierung MDR.
MDSAP Beratung – Ihr Weg zur erfolgreichen internationalen Zertifizierung
Das Medical Device Single Audit Program (MDSAP) ist ein weltweit anerkanntes Audit-Verfahren. Es ermöglicht Medizinprodukteherstellern, die regulatorischen Anforderungen mehrerer Länder mit nur einem Audit zu erfüllen. Australien, Brasilien, Kanada, Japan und die USA nehmen an diesem Programm teil. Alle verlangen die Einhaltung strenger Qualitätsstandards. Diese basieren auf der ISO 13485 sowie auf spezifischen nationalen Vorschriften.
Ein erfolgreiches MDSAP-Audit bringt zahlreiche Vorteile mit sich. Es reduziert den Aufwand für Hersteller, indem es die Anforderungen mehrerer Märkte in einem einzigen Prozess abdeckt. Einheitliche Auditverfahren sorgen für konsistente Ergebnisse und minimieren potenzielle Abweichungen. Gleichzeitig stärkt die Teilnahme am MDSAP die Produktsicherheit, da die Zusammenarbeit der Behörden eine frühzeitige Identifikation und Minimierung von Risiken ermöglicht. Diese Vorteile helfen Ihnen nicht nur, globale Märkte effizienter zu bedienen, sondern verschaffen Ihnen auch einen Wettbewerbsvorteil durch nachgewiesene Compliance.
Eine gründliche MDSAP Analyse ist der erste Schritt zur erfolgreichen MDSAP-Zertifizierung. Dabei geht es darum, Ihr bestehendes Qualitätsmanagementsystem (QMS) zu überprüfen und gezielt an die Anforderungen des MDSAP anzupassen. Genau hier setzen wir mit unserer MDSAP Beratung an.
Wir unterstützen Sie dabei:
- MDSAP GAP-Analyse: Wir analysieren Ihr bestehendes Qualitätsmanagementsystem (QMS) auf fehlende Themen des MDSAPs.
- Einführung MDSAP & individuelle QMS-Anpassung: Wir integrieren MDSAP-spezifische Anforderungen und stellen die ISO 13485-Konformität sicher.
- Praxisorientierte Beratung und Prozessoptimierung: Unser Team erstellt die erforderlichen QM-Unterlagen, identifiziert bestehende Lücken und optimiert Ihre Prozesse, um die Audit-Anforderungen zu erfüllen.
- MDSAP Audit Begleitung: Unsere Experten bereiten Sie gezielt auf das MDSAP-Audit vor und begleiten Sie durch den gesamten Zertifizierungsprozess – von der Planung bis zum Zertifikat.
Mit unserer MDSAP Beratung profitieren Sie von effizienteren Prozessen, reduzierten Kosten durch weniger redundante Audits, verbesserter Produktsicherheit und einem erleichterten Zugang zu den internationalen MDSAP-Märkten.
Kontaktieren Sie uns für eine unverbindliche Beratung und starten Sie Ihre Reise zur globalen Compliance mit dem Medical Device Single Audit Program (MDSAP).

FDA-Beratung zur 510(k)-Zulassung & Einführung QMSR
Hersteller von Medizinprodukten, die den US-Markt betreten möchten, benötigen eine fundierte FDA-Beratung, um die strengen Anforderungen der FDA-Zulassung zu erfüllen. Zwei zentrale Bestandteile sind die 510(k)-Zulassung und die Einführung QMSR (Quality Management System Regulation), die für die regulatorische Konformität entscheidend sind.
510(k)-Zulassung: Nachweis der wesentlichen Gleichwertigkeit
Die 510(k)-Zulassung fordert von Herstellern, aktiv nachzuweisen, dass ihr Produkt sicher und wirksam ist. Zusätzlich müssen sie belegen, dass ihr Produkt einem bereits zugelassenen Prädikatsprodukt entspricht. Dieses Verfahren betrifft vor allem Medizinprodukte der Klassen I und II.
Die FDA prüft dabei, ob das Produkt denselben Verwendungszweck erfüllt und vergleichbare technologische Eigenschaften besitzt. Gleichzeitig analysiert sie, ob keine neuen oder zusätzlichen Risiken die Sicherheit beeinträchtigen. Hersteller können eine FDA-Zulassung nur erreichen, wenn sie die sogenannte Substantial Equivalence (wesentliche Gleichwertigkeit) vollständig nachweisen.
Einführung QMSR: Die Grundlage für die FDA-Zulassung
Die Quality Management System Regulation (QMSR), die aus der überarbeiteten Quality System Regulation (QSR) der Verordnung 21 CFR Part 820 hervorgeht, harmonisiert die FDA-Vorgaben mit der internationalen Norm ISO 13485. Diese Anpassung unterstützt Hersteller dabei, globale Standards zu erfüllen und gleichzeitig die spezifischen Anforderungen der FDA umzusetzen. Die wichtigsten Neuerungen der QMSR:
- FDA-spezifische Ergänzungen: Die QMSR definiert detaillierte Vorgaben zur Design-Validierung, Risikoüberwachung und Änderungskontrolle.
- Kombinationsprodukte: Klare Regeln gelten für Produkte, die Medizinprodukte mit Arzneimitteln oder biologischen Komponenten kombinieren.
- Verstärkte Auditanforderungen: Die FDA hat ihre Erwartungen an die Überprüfung und Dokumentation interner Prozesse präzisiert, um eine konsistente Produktqualität zu gewährleisten.
Wir unterstützen Sie dabei:
Mit unserer spezialisierten FDA-Beratung unterstützen wir Sie umfassend bei der Einführung QMSR und der 510(k)-Zulassung:
- Individuelle QMS-Anpassung: Wir optimieren Ihr Qualitätsmanagement gemäß den Vorgaben der neuen Quality Management System Regulation (QMSR).
- Nachweis der Substantial Equivalence: Wir helfen Ihnen, ein geeignetes Prädikatsprodukt zu identifizieren und die erforderlichen Nachweise für die 510(k)-Zulassung zu erstellen.
- 510(k)-Zulassungsantrag: Wir übernehmen die Erstellung und Einreichung der Unterlagen für Ihre 510(k)-Zulassung.
- FDA-Inspektionen: Wir bereiten Sie optimal auf FDA-Prüfungen vor und stellen sicher, dass alle Anforderungen erfüllt sind.
Mit unserem Expertenwissen navigieren wir Sie sicher durch den FDA-Zulassungsprozess. Unsere FDA-Beratung hilft Ihnen, Ihre Produkte erfolgreich auf dem US-Markt zu etablieren.
IVDR Beratung – Experten für die IVDR Einführung und Zertifizierung
Die neue In-vitro-Diagnostika-Verordnung (IVDR), Abkürzung für die Verordnung (EU) 2017/746, regelt die Zulassung von In-vitro-Diagnostika in der Europäischen Union. Sie stellt sicher, dass Produkte höchsten Standards in Bezug auf Sicherheit, Qualität und Rückverfolgbarkeit entsprechen. Alle In-vitro-Diagnostika, die auf dem EU-Markt erhältlich sind, müssen eine umfassende IVDR Zertifizierung durchlaufen und die CE-Kennzeichnung erhalten. Diese bestätigt die Einhaltung der EU-Richtlinien und berechtigt zum Vertrieb innerhalb Europas. Die IVDR Prozesse umfassen eine detaillierte technische Dokumentation und die Einhaltung strenger regulatorischer Anforderungen. Die IVDR führt ein risikobasiertes Klassifizierungssystem ein, das Produkte in die Klassen A bis D einteilt.
Unser Expertenteam unterstützt Sie umfassend die neuen Anforderungen bei der Zulassung Ihrer In-vitro-Diagnostika gemäß IVDR zu bewältigen und begleitet Sie auf dem Weg zur vollständigen Konformität.
Wir unterstützen Sie dabei:
- IVDR Analyse: Wir prüfen Ihre Produktdokumentationen und identifizieren die erforderlichen Unterlagen und Nachweise für die IVDR-Zertifizierung. Durch eine sorgfältige Strukturierung Ihrer technischen Dokumentation schaffen wir eine solide Basis für den Zertifizierungsprozess.
- Maßgeschneiderte IVDR Einführung: Ein individueller Maßnahmenplan ist entscheidend für eine erfolgreiche IVDR Einführung. Unser Plan umfasst alle notwendigen Schritte zur Erreichung der IVDR-Konformität und berücksichtigt die spezifischen Anforderungen Ihrer In-vitro-Diagnostika.
- Begleitung bei der IVDR Umsetzung: Während der Implementierungsphase stehen wir Ihnen als erfahrener IVDR Berater zur Seite, um die Einhaltung aller Vorgaben zu gewährleisten und die erforderliche IVDR Zertifizierung zu erreichen.
- Erstellung der IVDR Dokumentation: Eine vollständige technische Dokumentation ist unerlässlich für die IVDR-Compliance. Wir unterstützen Sie bei der Erstellung und Pflege der notwendigen IVDR Prozesse, damit Sie optimal auf Audits vorbereitet sind.
Unsere IVDR Beratung – Ihr Partner für eine reibungslose IVDR Einführung
Beginnen Sie den Zulassungsprozess Ihrer In-vitro-Diagnostika mit Hopp+Flaig als verlässlichem Partner an Ihrer Seite. Wir kennen die Anforderungen der In-vitro-Diagnostika-Verordnung und begleiten Sie kompetent auf dem Weg zur CE-Kennzeichnung und erfolgreichen Platzierung Ihrer Produkte auf dem europäischen Markt.
Beratung zur Großhandelserlaubnis – Von der Antragstellung bis zur Zertifizierung
Für Unternehmen im Arzneimittelhandel ist die Erlangung einer Arzneimittel-Großhandelserlaubnis gemäß § 52a AMG unverzichtbar. Diese Erlaubnis weist nicht nur die Einhaltung aller gesetzlichen Anforderungen nach, sondern auch höchste Standards in Qualität und Sicherheit beim Umgang mit Arzneimitteln. Mit unserer umfassenden Beratung zur Großhandelserlaubnis für Apotheken führen wir Sie Schritt für Schritt durch den komplexen Prozess und sorgen dafür, dass Ihr Unternehmen erfolgreich agieren kann.
Voraussetzungen für die Großhandelserlaubnis
Um eine Arzneimittelgroßhandelserlaubnis zu erhalten, müssen Unternehmen eine Vielzahl von gesetzlichen Anforderungen erfüllen, die in der EU- „Leitlinien für die gute Vertriebspraxis von Humanarzneimitteln“ (GDP), Arzneimittel-Handelsverordnung (AM-HandelsV) und des Arzneimittelgesetzes (AMG) geregelt sind. Zu den wichtigsten Voraussetzungen zählen geeignete Lager- und Betriebsstätten, sachkundige Auslieferung, ein umfassendes Qualitätssicherungssystem, geschultes Personal sowie umfassende Rückverfolgbarkeit der Arzneimittel.
Wie wir Sie dabei unterstützen:
- Einführung Großhandelserlaubnis für Apotheken: Wir unterstützen Sie von der ersten Planung bis zur finalen Umsetzung der erforderlichen Maßnahmen.
- GAP-Analyse für Großhandelserlaubnis: Wir bewerten Ihre aktuelle Situation, identifizieren Schwachstellen und entwickeln maßgeschneiderte Strategien für eine erfolgreiche Antragstellung.
- Aufbau eines QMS: Wir erstellen oder optimieren Ihr Qualitätsmanagementsystem gemäß der den umfassenden gesetzlichen Anforderungen, ohne dabei Ihre individuelle Situation aus dem Auge zu verlieren.
- Vorlagen: Profitieren Sie von unseren erprobten Vorlagen und bewährten Hilfsmitteln, die Ihnen die Umsetzung der gesetzlichen Anforderungen erleichtern.
- Projektmanagement: Wir übernehmen die Koordination und Kommunikation mit den zuständigen Behörden, um einen reibungslosen Ablauf zu gewährleisten.
- Zertifizierung Großhandelserlaubnis für Apotheken: Ob interne Audits oder behördliche Überprüfungen – wir bereiten Sie optimal vor und begleiten Sie bei Bedarf, damit Sie den Prozess sicher und erfolgreich durchlaufen..
Mit unserer Expertise sparen Sie wertvolle Zeit. Wir sorgen für Ihren guten Eindruck bei den Behörden und einen professionellen Antrag auf Erteilung einer Arzneimittel-Großhandelserlaubnis. Kontaktieren Sie uns frühzeitig und profitieren Sie von unserem Know-how.
Ihr Nutzen in der Zusammenarbeit mit Hopp+Flaig
- kompetente Beratung, in die unsere langjährige Erfahrung einfließt
- intensive und lösungsorientierte Betreuung
- möglichst einfache und pragmatische Umsetzung der Normanforderungen
- möglichst einfache und pragmatische Lösungen
- reibungsloser und zügiger Projektverlauf durch Nutzung unserer bewährten Roadmaps
- Nutzung unserer bewährten Vorlagen für die notwendige Dokumentation
- kein interner Aufwand für den Aufbau von Spezialwissen
- weitere Reduzierung des internen Aufwands durch Übernahme von Spezialaufgaben wie z. B. Projektleitung, interne Audits oder Schulungen durch uns
- „gleiche Augenhöhe“ mit den Prüfern im Zertifizierungsprozess
- Immer wenn sinnvoll, Einsatz von Softwaretools
Motivation unserer Kunden
- Wunsch der Führungskräfte nach Ordnung, Struktur und verlässlichen Prozessabläufen
- Erlangen von Branchenqualifizierungen, ohne welche der Marktzutritt verwehrt bleibt
- Erhöhung der Rechtssicherheit
- Etablierung von Verbesserungsprozessen

